- Research
- Open access
- Published:
A deep phenotyping experience: up to date in management and diagnosis of Malan syndrome in a single center surveillance report
Orphanet Journal of Rare Diseases volume 17, Article number: 235 (2022)
Abstract
Background
Malan syndrome (MALNS) is a recently described ultrarare syndrome lacking guidelines for diagnosis, management and monitoring of evolutive complications. Less than 90 patients are reported in the literature and limited clinical information are available to assure a proper health surveillance.
Results
A multidisciplinary team with high expertise in MALNS has been launched at the “Ospedale Pediatrico Bambino Gesù”, Rome, Italy. Sixteen Italian MALNS individuals with molecular confirmed clinical diagnosis of MALNS were enrolled in the program. For all patients, 1-year surveillance in a dedicated outpatient Clinic was attained. The expert panel group enrolled 16 patients and performed a deep phenotyping analysis directed to clinically profiling the disorder and performing critical revision of previously reported individuals. Some evolutive complications were also assessed. Previously unappreciated features (e.g., high risk of bone fractures in childhood, neurological/neurovegetative symptoms, noise sensitivity and Chiari malformation type 1) requiring active surveillance were identified. A second case of neoplasm was recorded. No major cardiovascular anomalies were noticed. An accurate clinical description of 9 new MALNS cases was provided.
Conclusions
Deep phenotyping has provided a more accurate characterization of the main clinical features of MALNS and allows broadening the spectrum of disease. A minimal dataset of clinical evaluations and follow-up timeline has been proposed for proper management of patients affected by this ultrarare disorder.
Introduction
Malan syndrome (MALNS; MIM #614753), previously called “Sotos syndrome 2”, is an ultrarare overgrowth syndrome (OGS) characterized by postnatal overgrowth, macrocephaly, a distinctive facial gestalt, skeletal defects, developmental delay (DD)/intellectual disability (ID), and behavioral anomalies [1]. MALNS is caused by haploinsufficiency of the nuclear factor I X gene (NFIX; MIM #164005), due to either heterozygous chromosomal microdeletions involving the 19p13.2 region or loss-of-function (LoF) variants in the NFIX gene, the latter almost exclusively clustering within exon 2 [1, 2]. Based on the number of known affected individuals, the prevalence of this disease is estimated as 1/1.000.000 [3]. To date, less than 90 affected individuals have been reported [2, 4,5,6,7]. Interestingly, no clinically relevant genotype–phenotype correlations were observed comparing individuals with intragenic variants and gene deletions, except for a significantly higher frequency of epilepsy in individuals carrying NFIX microdeletions. This finding was explained as due to a contiguous gene effect [2]. MALNS is inherited in an autosomal dominant manner and is allelic to Marshall–Smith syndrome (MSS, MIM #602535), which is characterized by postnatal failure to thrive, short stature, dysostosis, post-natal failure to thrive, typical facial gestalt, respiratory compromise, and moderate to severe DD/ID [8,9,10]. In MSS, NFIX variants (frameshift variants or intragenic deletions confined to exons 6 to 7) escape nonsense-mediated mRNA decay (NMD), leading to anomalous proteins with an aberrant shared C-terminus, with preserved DNA binding and dimerization, resulting in a possible dominant-negative effect [1, 10]. By comparing the main features of MALNS and MSS, the two conditions have been characterized as clinically distinct and allelic entities despite some clinical overlap [2]. Recently, a more severe ID, impaired speech and language, less adaptive behavior skills, and reciprocal social interaction have been reported in MSS compared to MALNS [11]. The main features of MALNS according to previous literature and differential diagnosis are summarized in Table 1.
Results
-
(1)
General cohort assessment Nine males and seven females, with a median age of 13.4 years (IQR range: 3–25.6 years) were enrolled. Main clinical features are reported in Figs. 1, 2 and Table 2. Detailed profiling of facial features is provided in Fig. 3. The molecular characterization of the entire cohort is provided in Table 3. Fourteen individuals had NFIX pathogenic SV, and two carried NFIX microdeletion. Seven individuals had previously been reported [2]. Nine new individuals with MALNS have been described.
Fig. 1 
Facial feature panel of presently reported 16 individuals with Malan syndrome, with evolving facial appearance. Individuals are listed according to Table 2. Age in months (mths)/years (yrs) is reported below each picture. For detailed descriptions please see Tables and text
Fig. 2 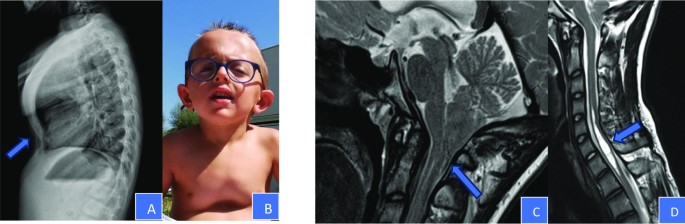
Musculoskeletal anomalies: A and B skeletal Xray and photo of case 16, that showed marked kyphosis with pectus carinatum in the upper half of the sternum and excavatum in the lower half. C and D MRI performed on case 1. The patient presented a Chiari malformation type I with a protrusion of cerebellar tonsils through the foramen magnum of 11 mm. Presence of syringomyelic cavity in the cervical spine, affecting the tract C5–C7
Table 2 Main features and molecular characterization of 16 Malan syndrome patients Fig. 3 
A Heatmap depicts common facial features for patients carrying NFIX deleterious variants by using the specific Human Phenotype Ontology (HPO) annotation (rows), which were retrieved from published studies and our cohort (columns). Phenotypic enrichment is shown according to the features’ recurrence labeled by the increment of color degree. The items with no features available were labeled white. B Is it possible to observe the numbers of patients presenting the specific feature (N°) and the percentage of the feature (%)
Table 3 Detailed molecular characterization of MALNS patients: seven patients were previously reported, and nine patients are new reports -
(2)
Cardiovascular assessment Cardiological evaluations with EcoCG were performed on all subjects and did not identify major cardiac anomalies or malformations nor dilatation of the aortic bulb or pulmonary artery. Mitral regurgitation (MR) was observed in 31% of cases. Patient 1 showed minimal pericardial effusion at EcoCG at the age of 13 years. Patient 13 showed a tricuspid aortic valve anomaly. Patient 14 presented with benign premature ventricular contractions (PVC) that were extensively investigated with a stress test and Holter ECG monitor. In this patient, mild aortic valve dysplasia was also reported.
-
(3)
Orthopedic assessment Musculoskeletal anomalies were frequently observed and required orthopedic evaluation and correction in some cases. Slender habitus was documented in all individuals, while long hands were observed in 63% and ligamentous hyperlaxity in 19%. Abnormal spine curvatures were observed in 75% of cases, 7 subjects had mild to severe scoliosis, 3 had hyper-kyphosis and 2 (Pts 6 and 8) with a combination of hyper-lordosis and scoliosis. One case (Pt 11) with severe scoliosis (Risser grade 3) underwent vertebral column arthrodesis at the age of 13 years old. Two additional patients (Pts 1 and 6) needed prescriptions of orthopedic insoles for the limb-length discrepancy. Patient 12 presented with severe levoscoliosis with a Cobb angle of 50 degrees, which was treated with a scoliosis brace. He is currently under orthopedic follow-up for the related high surgical risk. One case of hyper-kyphosis was surgically treated at 15 years (Pt 5). Structural deformities of the anterior thoracic wall were relatively common, with a frequency of 63%. Eight individuals showed different grades of pectus excavatum; one showed pectus carinatum, and one (Pt 16) had a severe mixed pattern of pectus carinatum and excavatum (Fig. 2A, B). The latter was investigated with an overnight pulse oximetry test which gave normal results and was evaluated by a thoracic surgeon and a pneumologist, who excluded a surgical treatment and/or noninvasive ventilation. Pes planus was recorded with high prevalence (70%). Two patients had been treated with bilateral subtalar arthrodesis (Pts 1 and 5). Diaphyseal fractures were observed in 5 subjects. Patient 4 presented with a tibia-fibula fracture at the age of 8 years. She underwent a dual-energy X-ray absorptiometry (DXA) at the age of 20 years that resulted normal. Patient 6 presented with three different episodes of tibial shaft fractures, both in right and left tibia, all occurring between 2 and 5 years, due to minor trauma. Patient 8 showed bilateral tibial shaft fractures. Patient 14 presented with unilateral tibial shaft fracture and underwent a DXA exam at the age of 15 years old that showed mild femoral osteopenia with a Z-score of − 2.1. She started supplementation with vitamin D3 and repeated the DXA at the age of 17 years that showed a rise in bone mineral density of 4.5% compared to the previous exam. Patient 15 presented with a unilateral tibial shaft fracture. Tibial fibrous cortical defect of 17 mm (Pt 1), bullet-shape epiphysis (Pt 4) and hyperostosis of the fifth finger of the hands, bilaterally (Pt 11) were also noticed.
-
(4)
Neurological assessment Neonatal hypotonia was observed in 50% of our cohort. The EEG showed isolated unspecific anomalies in 8 individuals (50%), 7 of them carrying an intragenic NFIX variant and 1 having a NFIX microdeletion encompassing the CACNA1A gene. Patient 3 carrying an intragenic NFIX pathogenic variant was treated for EEG anomalies alone (detected in the second year of life) with valproic acid for 1 year until the normalization of EEG pattern. Two patients presented with epilepsy (Pts 14 and 16). Patient 14 experienced an episode of seizure at the age of 8 years with a positive EEG. She started a treatment with Topiramate that was interrupted at the age of 15 years due to the normalization of the EEG pattern. Patient 16 experienced a first episode of tonic–clonic seizures with loss of consciousness at the age of 6.5 years with a positive EEG. For this reason, he started antiepileptic therapy with valproic acid. Due to the relapse of symptoms with a new critical episode, drug therapy was modified.
All individuals were screened for brain malformation by MRI. Ventriculomegaly (50%) and different degrees of CCH (50%) were reported. CM1 was observed in 38% of cases. Patient 1 presented with a combination of CM1 and syringomyelia, a well-known association [15] (Fig. 2C, D) with a syringomyelic cavity in the cervical spine (C5-C7 segments). He performed somatosensory evoked potentials (SEPP), which resulted negative, and was evaluated by a neurosurgeon who suggested a 6-month follow-up with MRI. Patient 9 had a multicystic pineal gland and two leptomeningeal cysts at MRI; in addition, this subject showed bilateral sensorineural hearing loss previously treated with cochlear implant as a previously described sign [2]. Other observations were dysgiria and megalencephaly associated with dilatation of central canal of the spinal cord (Pt 8), punctiform area of gliosis in deep white brain matter, likely due to perinatal distress (Pt 13), septum pellucidum cysts (Pt 14) and dystopic neurohypophysis (Pt 16). Neurovegetative anomalies and gait disturbances were also noticed. Patient 1suffered from postural fainting and episodes of vomiting; patient 7 suffered from frequent falls; patient 15had bouts of dizziness, nausea, vomiting, and ataxic gait treated with acetazolamide with amelioration in symptoms.
-
(5)
Neuropsychiatric assessment All individuals had ID. Clinical diagnosis of mild ID was made only for one individual, while the other showed moderate or severe degree of ID. Due to young age, patient 10 received a diagnosis of developmental delay (DD). Ninety percent of individuals showed language impairment of various degrees, generally moderate or severe, in some cases with absence of speech. In general, expressive language was more impaired than receptive language. Almost all individuals presented with behavioral problems, such as anxiety and, to a lesser extent, autistic-like behaviour. Patient 6 presented with an increase in anxiety and self-aggressive behavior associated with a sleep disorder at the age of 24 years; for this reason, she started drug therapy with Mirtazapine, with amelioration in symptoms. Diminished visuomotor integration abilities were a frequent finding observed in virtually all patients. Occasionally we also suspected oculomotor apraxia in some MALNS individuals, but their neurological assessment has been separately performed and results will be provided in a separate manuscript (Alfieri et al., submitted). High sensitivity to noises was referred in 13 (81%) patients, in three cases associated with photophobia (Pts 1, 5 and 14) (not reported in Table 1). Three patients were recommended to use colored lenses (Pt 5, 14 and 16), with amelioration in photophobia and social interactions.
-
(6)
Ophthalmological assessment A high frequency of refractive disorders with a prevalence of 80% was observed. Four individuals had hypermetropia and nine had myopia. Three of them also presented with astigmatism. Other frequent findings were esotropia, (56%), strabismus (63%) and nystagmus (31%). Patient 11 underwent surgical correction for esotropia at 16 years of age and performed several visual evoked potentials (VEPs) and electroretinogram tests that showed altered cortical retinal transmission with normal retinal function. Anomalous VEP patterns were evidenced in patient 1, as well. Blue sclerae were observed in 69%. Bilateral polar cataracts in two cases (Pts 1 and 5) were reported. Four patients (Pts 4, 5, 7 and 9), all previously reported in the literature, presented with ONH while 4 patients (two previously reported Pts 7 and 9 and two new report Pts 1 and 4), presented with optic disk pallor.
-
(7)
Orodental assessment High arched palate with dental overcrowding was observed in 56% of cases. Different types of dental malocclusion (both class II and III of Angel’s classification) were observed, together with an open bite in 44%. Caries were observed in 38%. Oral apraxia and sialorrhea were noticed with an occurrence of 31%.
-
(8)
Other relevant clinical findings BMI and BMI-SDS were assessed in all participants resulting in a median BMI of 16.5 (interquartile (IQR): 13.2–22.5) in males and 17.4(IQR: 12–22.3) in females, with 6 individuals presenting with severe thinness (considered as BMI-SDS < 2SD or < 3rd percentile) (Additional file 1: Fig. S1). Two of them underwent nutrition assessment by an expert dietitian that confirmed normal intake of micro and macro-nutrients for age. Hepatomegaly was found in 25% of cases, a finding previously unappreciated. Constipation was a frequent observation with a prevalence of 50%. Two individuals underwent orchidopessis for bilateral cryptorchidism (Pts 2 and 3), and one (Pt 13) had surgical correction of IV grade hydronephrosis due to pyeloureteral junction stenosis associated with oligohydramnios as a previously described sign [2] and surgical correction for bilateral inguinal hernia. Patient 8 showed clitoral hypertrophy, with P1B1 Tanner stage. Frequent infections have been observed in patient 9 and patient 7, the latter also showing hypogammaglobulinemia. Patient 8 had a diagnosis of Wilms' Tumor of the left kidney with subcentimetric pulmonary metastasis at 5 years. He underwent chemotherapy and subsequently nephrectomy, according to TW2003 protocol for Wilms’ Tumor treatment developed by Associazione Italiana Ematologia e Oncologia Pediatrica (AIEOP) [16]. He is off therapy since December 2018.



Discussion
Figure 4 and Additional file 1: Table S1 report the comparison between our single-center cohort and the previously reported multicenter cohort [2]. On the whole, we found significant differences in the occurrence of many signs with respect to the previous cohort data and added ten new features to the spectrum of presentation in MALNS (Additional file 1: Table S1 shows a comparison of the most significant signs/features). These apparent discrepancies between the two series may be easily explained. Some features (i.e., altered EEG patterns identified through systematic EEG recording, neurovegetative signs, Chiari malformation, subtle corpus callosum hypoplasia, esotropia, and gastrointestinal signs) were assessed in all patients or asked to the caregivers, based on the authors’ previous experience (M.M. and M.P). Other signs (i.e., long bone fractures, and transiently reduced bone density) emerged after an accurate personal history evaluation that was possible by close periodic follow up. Musculoskeletal anomalies scored with significant differences in almost all features. Interestingly, a higher prevalence of abnormal spine curvatures was identified allowing a subclassification of spinal curvature anomalies. Pes Planus is a new but persistent observation surgically treated in two cases. Long bone fractures were evidenced in five individuals. The presence of diaphyseal bone fractures and reduced bone mineral density at DXA in one of them might be indicative of a higher risk of osteopenia/osteoporosis, which could overlap with a similar finding in MSS patients, who typically present with marked osteopenia [2]; however, the control DXA in this patient at the age of 17 was normal as the DXA performed at the age of 20 in one other of the previously fractured patients (Pt 4). Further studies are needed to investigate bone mineral density by performing DXA exams in post-pubertal MALNS individuals.

Compared frequencies of main Malan syndrome features between our cohort and Priolo et al., 2018 cohort. A Neurological features and Brain MRI. B Musculoskeletal features. C Ophthalmological features. D Other findings. Ten new features were reported among our cohort, indicated as New Report. Detailed information was reported in the text. NR New report
Due to the presence of slender habitus and the possible presence of a systemic score ≥ 7 of the revised Ghent criteria (e.g., scoliosis, wrist and thumb sign, pectus carinatum, myopia or mitral valve prolapse) [17]. Marfan syndrome (MIM 1547009) should be considered as a possible differential diagnosis but it can be easily ruled out by the constant presence in MALNS patients of ID and the absence of typical Marfan facial gestalt or aortic root enlargement.
MALNS may be misdiagnosed as a syndrome with marfanoid habitus and ID (e.g. homocystinuria [MIM 236200], Snyder–Robinson syndrome [MIM 309583], marfanoid mental retardation syndrome [MIM 248770], Lujan–Fryns syndrome [MIM 309520]), though the facial gestalt and clinical presentation of these disorders significantly differ from MALNS. Molecular confirmation is required to assess proper diagnosis.
Epilepsy and isolated EEG anomalies were also more frequently observed. Individuals with NFIX intragenic variants were prone to develop aspecific EEG anomalies, which did not require anti-epileptic therapy. Brain MRI was performed in all individuals documenting a higher prevalence of structural brain anomalies (wide ventricles, CCH and CM1) confronted with the previous series. Recurring neurovegetative signs also emerged as a relevant clinical issue in MALNS. The previous series reported 1 patient with postural fainting. By retrospectively reviewing all other MALNS individuals affected with 19p13.2 microdeletions involving NFIX, three additional cases with cyclic vomiting, dizziness, nausea, and sporadic ataxic gait were recognized [18, 19]. It has been hypothesized that co-occurring CACNA1A deletion could be an associated cause in the pathogenesis of these signs [19]. Neurovegetative signs and gait disturbances have also been occasionally described in CM1 as brainstem/cerebellar-related symptoms [20, 21]. By identifying the presence of CM1 in some of the 7 MALNS individuals presenting with neurovegetative symptoms, we speculate that mixed mechanisms likely trigger them. Three out seven patients had a co-occurring CACNA1A deletion, one subject had a microdeletion not involving CACNA1A but was affected with CM1 and syringomyelia and one subject (Pt 1) carrying a pathogenetic NFIX variant had CM1 with syringomyelia (Additional file 1: Table S2). Two other cases had neither CACNA1A deletion nor CM1. We concluded that for the first one, previously described [2], postural hypotension was a too general sign to be considered a true neurovegetative one. Patient 7 in the present series showed unsteady gait and frequent falls, but never showed neither neurovegetative symptoms such as vomit, nausea, or abdominal pain nor neurologic symptoms such as dizziness or vertigo. So, we can hypothesize that her clinical picture could be explained by the frequent finding of diminished visuomotor integration abilities within the MALNS population.
Neuropsychiatric and behavior evaluation revealed moderate to severe ID in almost all patients with the exception of one who was diagnosed with mild DD (Pt10). Behavioural problems with peculiar anxious profile have been evidenced in more than 50%. Diminished visuomotor integration abilities seem to be a neuropsychiatric hallmark in MALNS such as noise sensitivity and photophobia. Colored lenses are currently used by autistic patients to reduce light stimuli triggers and improves social tasks [22]. Three patients of the present series (Pt 5, 14 and 16) successfully adopted the same strategy with the benefice of reducing anxiety outbursts. Due to the complexity of the methodology of assessment, neuropsychiatric results will be provided in a separate manuscript (Alfieri et al., submitted).
A multicystic pineal gland and meningeal cysts have also been recorded. Both signs have been previously recorded in MALNS [2, 18] although these are quite common MRI finding in general population [23, 24]. Nonetheless, their progression should be monitored to prevent associated neurologic symptoms.
A comparable frequency of refractive disorders with respect to previous reports has been observed. On the other hand, a higher frequency of strabismus and nystagmus has been recorded. Of note, esotropia had a prevalence of 56%; bilateral polar cataract (with a probable congenital etiology) was detected in patients 1 and 5 during the current evaluation although they were previously described [2]. Optic Nerve Hypoplasia (ONH) and Optic Disk Pallor (ODP) were evidenced in 25% of cases, respectively.
Cardiovascular diseases had been previously claimed as a possible main feature of MALNS due to the frequent observation of Marfanoid habitus [2]. None of the individuals in the present series showed aortic bulb or pulmonary artery dilatation. Minor anomalies such as low-grade mitral regurgitation (MR) in 31% have been observed. In a large cohort study on the adult population (The Framingham Heart Study) MR was detectable at echocardiographic examinations in a vast control population, resulting in trace or mild MR [25]. We might conclude that these minor anomalies are not sufficient to classify MALNS as a condition predisposing to cardiovascular disease and/or heart anomalies.
Visceromegaly is a well-known association in OGS [26]. Isolated hepatomegaly was observed in 25% of MALNS patients here described. Different degrees of constipation were observed in 50% of our cohort, in some cases requiring pharmacological therapy with polyethylene glycol. Functional constipation is a frequent problem in patients with ID or DD [27]. Nonetheless, due to the high occurrence in MALNS population and the consequent impact on the quality of life, great attention should be paid to this complication.
Six individuals with a BMI-SDS < 2SDS have been observed, indicative of severe thinness (Additional file 1: Fig. S1). Interestingly, NFIX-null mice are characterized by a reduced body size and inability to fully extend their limbs, suggesting a possible muscular phenotype [28]. NFIX drives transcriptional changes from embryonic to fetal myogenesis by specifically activating fetal genes [29]. Mice lacking NFIX show reduced myofiber cross sectional area. NFIX also acts through an inhibitory mechanism at the promoter of the gene that encodes for myostatin, a TGF- β family member with anti-myogenic properties [30]. This finding is consistent with the hypothesis that MALNS individuals could show a reduced muscular mass with the final consequence of the inability to gain weight, despite adequate nutrient intakes. The Marfanoid-progeroid-lipodystrophy syndrome (MPLS) (MIM 616914), a newly recognized fibrillinopathy caused by pathogenic dominant negative variants clustering in FBN1 exon 65 [31], is also characterized by extremely low BMI with reduced subcutaneous fat. Aberrant activation of the TGF-β signaling pathway in a SMAD-dependent manner has been demonstrated in a subset of individuals affected with MPLS. The TGF-β plays a role in fat metabolism with an inhibitory action on human adipose tissue development [32]. This evidence suggests that MALNS individuals may present with a reduced BMI caused by either muscular or fat involvement likely through the same TGF-β signaling pathway dysregulation. Further studies are needed to confirm the role of NFIX LoF in TGF-β signaling regulation.
A higher frequency of high arched palate with overcrowded teeth has been observed, as well. Accurate orodental evaluation at diagnosis and then strict follow up is highly recommended.
The second case of cancer in MALNS was recorded (Pt 8). OGS are usually associated with an increased risk of tumors with only slightly increased prevalence in most OGS when compared with the general population [33]. Wilms tumor is an embryonic tumor quite represented among OGS although with significant different prevalence in risk [33]. To date, 2 individuals (Pt 8 of the present report and Pt 43 from [2]) with MALNS with different types of neoplasms have been reported in the literature with and overall prevalence of 2.2% thus including MALNS in the same low risk group as SS and WVS with a low likelihood of developing cancer [33, 34]. We are aware that MALNS cohort is too small to calculate an absolute risk so we might suggest that routine surveillance is not recommended considering psychosocial implications for the affected individuals and their families, as well as the cost of an active instrumental cancer surveillance.
As additional information we reported two MALNS individuals conceived via Assisted Reproductive Technology (ART), one previously described (Pt 1) and one new report (Pt 14), and they add to another one previously described (Pt 21 from [2]).
A proposal for diagnosis, management and follow up of the main complication in Malan syndrome
Due to the recent description of MALNS and its rarity, there are no current guidelines to help clinicians in diagnosis, management and follow up. Here we propose a minimal dataset of clinical evaluation to be applied to MALNS individuals to precociously identify possible complications and assure proper management and follow up (Table 4), based on previous reports and the results of our experience. We strongly suggest auxologic (with periodic BMI evaluations), orthopedic, ophthalmologic, and neurological evaluations and strict follow up. Caregivers should be properly trained to recognize subtle neurological signs (seizures and/or neurovegetative signs) and should be informed of higher recurrence of pathologic fractures of long bones in their children. DXA assessment and vitamin D dosage may be recommended during puberty to eventually provide adequate supplementation. Neuropsychiatric and behavior assessments need a separate dataset and recommendations due to their detailed phenotyping (Alfieri et al. in preparation).
MALNS is inherited as an autosomal dominant condition so each individual with MALNS has a 50% chance of transmitting the disorder. To the best of our knowledge, reproductive fitness in MALNS is extremely low, due to the severe ID. Currently, no MALNS offspring have been reported in the literature. Most individuals diagnosed with MALNS carry a de novo genetic alteration (either microdeletion or SV). However, 6 previous individuals from 3 unrelated families with recurrence of disease due to either gonadal or parental mosaicism have been described [2, 6, 7]. Recommendations for the evaluation of parents of an individual with MALNS include appropriate genomic/genetic testing to exclude parental mosaicism. If the genetic alteration cannot be detected in either parent, it’s highly probable that the proband has a de novo event. However, gonadal mosaicism cannot be excluded. In that case, prenatal genetic counselling should be performed to inform parents about their residual low recurrence risk, and prenatal diagnosis could be offered. Due to the reporting of three MALNS individuals conceived by ART and with an apparently de novo pathogenic alteration, great attention should be paid in these families if other fetuses conceived by ART present as LGA.
Conclusions
Deep phenotyping in MALNS has helped to identify a more accurate occurrence of main features and has allowed us to expand the spectrum of signs and symptoms characterizing this ultrarare disorder. The high expertise of the team involved in all the evaluations contributed to promptly identify main and critical features to eventually ensure uniformity in the treatment of complications. A minimal dataset of clinical evaluations and follow-up timeline has been proposed for proper management in MALNS. A more detailed study on behavioral disturbances is in preparation. Further studies are needed to confirm our findings in a larger cohort of MALNS individuals.
Materials and methods
Population and molecular diagnosis
Sixteen genetically confirmed Italian MALNS individuals were recruited from the outpatient clinic of the “Clinical Genetic and Rare Diseases” Unit, Ospedale Pediatrico Bambino Gesù (Rome, Italy), from September 2020 to November 2021 and enrolled in the study. Molecular diagnosis was attained by clinical exome sequencing (CES) or comparative genomic hybridization/SNP array, which identified intragenic NFIX pathogenic variants or NFIX microdeletions in the frame of diagnostic testing.
Study design
All the participants underwent a multidisciplinary assessment based on the previous description of main features and complications, as previously reported [2]. They were firstly evaluated by an expert panel group (MM, MP, PA, MC), then monitored with a multistep assessment involving several specialists (e.g., neurologists, neurosurgeons, ophthalmologists, orthopedic surgeons), depending on the specific individual clinical history or needs. They underwent: (I) a thorough pediatric evaluation (MM) with the co-presence of an experienced clinical geneticist (MP); (II) a neuropsychiatric evaluation to assess cognitive, neuropsychological and psychopathological comorbidities profile characteristics (PA and CC); (III) a cardiological evaluation (MC) with echocardiogram to exclude possible heart malformation and/or aortic root or pulmonary artery dilatations. Depending on the clinical conditions of each subject, orthopedic evaluation was performed by radiological imaging to investigate skeletal anomalies (e.g., scoliosis, kyphosis/lordosis, and pectus excavatum), and neurological/neurosurgical evaluation, with CNS imaging directed to detect possible brain anomalies (e.g., Chiari malformation type 1 (CM1), wide ventricles, and corpus callosum hypoplasia (CCH).
Body mass index (BMI) and BMI-standard deviation score (SDS) were calculated according to the World Health Organization (WHO) guidelines [35] while BMI charts for boys and girls were drafted in accordance with Centers for Disease Control and Prevention (CDC) [36].
Ethics
All procedures were in accordance with the ethical standards of the institutional and/or national research committee and the 1964 Helsinki declaration and its later amendments or comparable ethical standards. All information has been collected during the clinical assessment in Bambino Gesù Children’s Hospital, after obtainment of informed consent and registered anonymously in the electronic medical record.
Availability of data and materials
All data on our cohort were collected during medical assessment in Bambino Gesù Children’s Hospital, after obtainment of informed consent.
Abbreviations
- BMI:
-
Body Mass Index
- CCH:
-
Corpus callosum hypoplasia
- CM1:
-
Chiari malformation type 1
- DD:
-
Developmental delay
- DXA:
-
Dual-energy X-ray absorptiometry
- EcoCG:
-
Echocardiogram
- EEG:
-
Electroencephalogram
- ID:
-
Intellectual disability
- IQR:
-
Interquartile
- LGA:
-
Large for gestational age
- LoF:
-
Loss-of-function
- MALNS:
-
Malan syndrome
- MSS:
-
Marshall–Smith syndrome
- MR:
-
Mitral regurgitation
- MRI:
-
Magnetic resonance imaging
- NFIX :
-
Nuclear factor I X
- OGS:
-
Overgrowth syndromes
- PVC:
-
Premature ventricular contractions
- SDS:
-
Standard deviations score
- SEPP:
-
Somatosensory evoked potentials
- SS:
-
Sotos syndrome
- SV:
-
Single variant
- TGF-β:
-
Transforming growth factor-β
- US:
-
UltraSound
- URD:
-
Ultra rare disease
- WHO:
-
World Health Organization
- WVS:
-
Weaver syndrome
References
Malan V, Rajan D, Thomas S, et al. Distinct effects of allelic NFIX mutations on nonsense-mediated mRNA decay engender either a Sotos-like or a Marshall–Smith syndrome. Am J Hum Genet. 2010;87(2):189–98.
Priolo M, Schanze D, Tatton-Brown K, et al. Further delineation of Malan syndrome. Hum Mutat. 2018;39(9):1226–37.
Orphanet-The portal for rare diseases and orphan drugs-Malan Syndrome https://www.orpha.net/consor/cgibin/Disease_Search.php?lng=EN&data_id=23101&Disease_Disease_Search_diseaseGroup=malan&Disease_Disease_Search_diseaseType=Pat&Disease(s)/groupofdiseases=Malan-overgrowth-syndrome&title=Malanovergrowthsyndrome&search=Disease_Search_Simple
Bellucco FT, de Mello CB, Meloni VA, Melaragno MI. Malan syndrome in a patient with 19p13.2p13.12 deletion encompassing NFIX and CACNA1A genes: case report and review of the literature. Mol Genet Genomic Med. 2019;7(12):e997.
Tabata K, Iida A, Takeshita E, et al. A novel pathogenic NFIX variant in a Malan syndrome patient associated with hindbrain overcrowding. J Neurol Sci. 2020;412:116758.
Hancarova M, Havlovicova M, Putzova M, et al. Parental gonadal but not somatic mosaicism leading to de novo NFIX variants shared by two brothers with Malan syndrome. Am J Med Genet A. 2019;179(10):2119–23.
Sihombing NRB, Winarni TI, van Bokhoven H, van der Burgt I, de Leeuw N, Faradz SMH. Pathogenic variant in NFIX gene affecting three sisters due to paternal mosaicism. Am J Med Genet A. 2020;182(11):2731–6.
Martinez F, Marín-Reina P, Sanchis-Calvo A, et al. Novel mutations of NFIX gene causing Marshall–Smith syndrome or Sotos-like syndrome: one gene, two phenotypes. Pediatr Res. 2015;78(5):533–9.
Shaw AC, van Balkom ID, Bauer M, et al. Phenotype and natural history in Marshall–Smith syndrome. Am J Med Genet A. 2010;152A(11):2714–26.
Schanze D, Neubauer D, Cormier-Daire V, et al. Deletions in the 3’ part of the NFIX gene including a recurrent Alu-mediated deletion of exon 6 and 7 account for previously unexplained cases of Marshall–Smith syndrome. Hum Mutat. 2014;35(9):1092–100.
Mulder PA, van Balkom IDC, Landlust AM, et al. Development, behaviour and sensory processing in Marshall–Smith syndrome and Malan syndrome: phenotype comparison in two related syndromes. J Intellect Disabil Res. 2020;64(12):956–69.
Huisman S, Mulder P, Kuijk J, et al. Self-injurious behavior. Neurosci Biobehav Rev. 2018;84:483–91.
Nimmakayalu M, Horton VK, Darbro B, et al. Apparent germline mosaicism for a novel 19p13.13 deletion disrupting NFIX and CACNA1A. Am J Med Genet A. 2013;161A(5):1105–9.
Oshima T, Hara H, Takeda N, et al. A novel mutation of NFIX causes Sotos-like syndrome (Malan syndrome) complicated with thoracic aortic aneurysm and dissection. Hum Genome Var. 2017;4:17022.
Saletti V, Farinotti M, Peretta P, Massimi L, Ciaramitaro P, Motta S, Solari A, Valentini LG. The management of Chiari malformation type 1 and syringomyelia in children: a review of the literature. Neurol Sci. 2021;42(12):4965–95.
Associazione Italiana Ematologia Oncologia Pediatrica (AIEOP)—Italian Guidelines for Wilms’ Tumor treatment. https://www.aieop.org/web/operatori-sanitari/gruppi-di-lavoro/tumore-di-wilms/.
Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47(7):476–85. https://doi.org/10.1136/jmg.2009.072785.
Dolan M, Mendelsohn NJ, Pierpont ME, Schimmenti LA, Berry SA, Hirsch B. A novel microdeletion/microduplication syndrome of 19p13.13. Genet Med. 2010;12(8):503–11.
Klaassens M, Morrogh D, Rosser EM, et al. Malan syndrome: Sotos-like overgrowth with de novo NFIX sequence variants and deletions in six new patients and a review of the literature. Eur J Hum Genet. 2015;23(5):610–5.
Di Cristofori A, Fusi L, Gomitoni A, Grampa G, Bersano A, Lombardia GENS collaborators. R583Q CACNA1A variant in SHM1 and ataxia: case report and literature update. J Headache Pain. 2012;13(5):419–23.
McClugage SG, Oakes WJ. The Chiari I malformation. J Neurosurg Pediatr. 2019;24(3):217–26.
Ludlow AK, Giannadou A, Franklin A, Allen PM, Simmons DR, Wilkins AJ. The possible use of precision tinted lenses to improve social cognition in children with autism spectrum disorders. Vision Res. 2020;170:53–9.
Cauley KA, Linnell GJ, Braff SP, Filippi CG. Serial follow-up MRI of indeterminate cystic lesions of the pineal region: experience at a rural tertiary care referral center. AJR Am J Roentgenol. 2009;193(2):533–7.
Epelman M, Daneman A, Blaser SI, et al. Differential diagnosis of intracranial cystic lesions at head US: correlation with CT and MR imaging. Radiographics. 2006;26(1):173–96.
Singh JP, Evans JC, Levy D, et al. Prevalence and clinical determinants of mitral, tricuspid, and aortic regurgitation (the Framingham Heart Study) [published correction appears in Am J Cardiol 1999 Nov 1;84(9):1143]. Am J Cardiol. 1999;83(6):897–902.
Manor J, Lalani SR. Overgrowth syndromes—evaluation, diagnosis, and management [published correction appears in Front Pediatr. 2020 Dec 23;8:624141]. Front Pediatr. 2020;8:574857.
Tabbers MM, DiLorenzo C, Berger MY, et al. Evaluation and treatment of functional constipation in infants and children: evidence-based recommendations from ESPGHAN and NASPGHAN. J Pediatr Gastroenterol Nutr. 2014;58(2):258–74.
Driller K, Pagenstecher A, Uhl M, et al. Nuclear factor I X deficiency causes brain malformation and severe skeletal defects. Mol Cell Biol. 2007;27(10):3855–67.
Messina G, Biressi S, Monteverde S, et al. Nfix regulates fetal-specific transcription in developing skeletal muscle. Cell. 2010;140(4):554–66.
Rossi G, Antonini S, Bonfanti C, et al. Nfix regulates temporal progression of muscle regeneration through modulation of myostatin expression. Cell Rep. 2016;14(9):2238–49.
Lin M, Liu Z, Liu G, et al. Genetic and molecular mechanism for distinct clinical phenotypes conveyed by allelic truncating mutations implicated in FBN1. Mol Genet Genomic Med. 2020;8(1):e1023. https://doi.org/10.1002/mgg3.1023.
Petruschke T, Röhrig K, Hauner H. Transforming growth factor beta (TGF-beta) inhibits the differentiation of human adipocyte precursor cells in primary culture. Int J Obes Relat Metab Disord. 1994;18(8):532–6.
Brioude F, Toutain A, Giabicani E, Cottereau E, Cormier-Daire V, Netchine I. Overgrowth syndromes—clinical and molecular aspects and tumour risk. Nat Rev Endocrinol. 2019;15(5):299–311.
Villani A, Greer M-L, Kalish JM, et al. Recommendations for cancer surveillance in individuals with RASopathies and other rare genetic conditions with increased cancer risk. Clin Cancer Res. 2017;23(12):e83–90.
World Health Organization (WHO)—Child Growth Standards—Body Mass Index for age. https://www.who.int/tools/child-growth-standards/software.
BMI charts according to Centers for Disease Control and Prevention (CDC). https://www.cdc.gov/healthyweight/assessing/bmi/childrens_bmi/about_childrens_bmi.html.
Acknowledgements
We thank the patients and their families for their generous participation to the study.
Funding
No funding was secured for this study. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Author information
Authors and Affiliations
Contributions
Dr. MM, MP, FMP and PA conceptualized and designed the article, collected data, drafted the initial manuscript, and reviewed and revised the final manuscript. Dr. DV, MVG, CF, CC, ZM, MA, MS and MC conceptualized and designed the article, drafted, and wrote the manuscript, and reviewed and revised the final manuscript. Drs. MT, CM and AB critically reviewed the manuscript for important intellectual content. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All procedures performed in this study involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Consent for publication
Data were saved on an electronic medical database.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Supplementary figures and tables.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Macchiaiolo, M., Panfili, F.M., Vecchio, D. et al. A deep phenotyping experience: up to date in management and diagnosis of Malan syndrome in a single center surveillance report. Orphanet J Rare Dis 17, 235 (2022). https://doi.org/10.1186/s13023-022-02384-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-022-02384-9