- Review
- Open access
- Published:
Systemic therapy of Cushing’s syndrome
Orphanet Journal of Rare Diseases volume 9, Article number: 122 (2014)
Abstract
Cushing’s disease (CD) in a stricter sense derives from pathologic adrenocorticotropic hormone (ACTH) secretion usually triggered by micro- or macroadenoma of the pituitary gland. It is, thus, a form of secondary hypercortisolism. In contrast, Cushing’s syndrome (CS) describes the complexity of clinical consequences triggered by excessive cortisol blood levels over extended periods of time irrespective of their origin. CS is a rare disease according to the European orphan regulation affecting not more than 5/10,000 persons in Europe. CD most commonly affects adults aged 20–50 years with a marked female preponderance (1:5 ratio of male vs. female). Patient presentation and clinical symptoms substantially vary depending on duration and plasma levels of cortisol. In 80% of cases CS is ACTH-dependent and in 20% of cases it is ACTH-independent, respectively. Endogenous CS usually is a result of a pituitary tumor. Clinical manifestation of CS, apart from corticotropin-releasing hormone (CRH-), ACTH-, and cortisol-producing (malign and benign) tumors may also be by exogenous glucocorticoid intake. Diagnosis of hypercortisolism (irrespective of its origin) comprises the following: Complete blood count including serum electrolytes, blood sugar etc., urinary free cortisol (UFC) from 24 h-urine sampling and circadian profile of plasma cortisol, plasma ACTH, dehydroepiandrosterone, testosterone itself, and urine steroid profile, Low-Dose-Dexamethasone-Test, High-Dose-Dexamethasone-Test, after endocrine diagnostic tests: magnetic resonance imaging (MRI), ultra-sound, computer tomography (CT) and other localization diagnostics. First-line therapy is trans-sphenoidal surgery (TSS) of the pituitary adenoma (in case of ACTH-producing tumors). In patients not amenable for surgery radiotherapy remains an option. Pharmacological therapy applies when these two options are not amenable or refused. In cases when pharmacological therapy becomes necessary, Pasireotide should be used in first-line in CD. CS patients are at an overall 4-fold higher mortality rate than age- and gender-matched subjects in the general population. The following article describes the most prominent substances used for clinical management of CS and gives a systematic overview of safety profiles, pharmacokinetic (PK)-parameters, and regulatory framework.
Introduction
Both, Cushing’s disease (CD) and Cushing’s syndrome (CS), are rare diseases characterized by high cortisol blood levels and impairment of circadian oscillation [1]. CD derives from pathologic adrenocorticotropic hormone (ACTH) secretion usually triggered by micro- or macroadenoma of the pituitary gland [2]. It is, thus, a form of secondary hypercortisolism. In contrast, CS describes the complexity of clinical consequences triggered by excessive cortisol blood levels over extended periods of time irrespective of their origin [3]. Clinical manifestation of CS, apart from corticotropin-releasing hormone (CRH-), ACTH-, and cortisol-producing (malign and benign) tumors may also be by exogenous glucocorticoid intake (Figure 1B). Patient presentation and clinical symptoms substantially vary depending on duration and plasma levels of cortisol. In 80% of cases CS is ACTH-dependent and in 20% of cases is ACTH-independent, respectively [4]. Clinical differentiation with respect to the onset of a certain treatment algorithm is by ACTH dependency and independency, respectively:
ACTH-dependent
-
➢ Pituitary adenoma (CD in strict sense) ~ 70% [5]
-
➢ Ectopic secretion of ACTH by non-pituitary tumors ~ 15% (i.e. neuroendocrine tumors such as small-cell lung cancer (SCLC), carcinoid tumors, and medullary carcinoma of the thyroid) [6]
-
➢ Ectopic secretion of CRH by non-hypothalamic tumors causing pituitary hypersecretion of ACTH < 1% [7]
-
➢ Iatrogenic or factitious CS due to administration of exogenous ACTH < 1%
ACTH-independent
-
➢ Adrenocortical adenomas and carcinomas ~ 20% [8]
-
➢ Primary pigmented nodular adrenocortical disease < 1% [9]
-
➢ Bilateral ACTH-independent adrenal hyperplasia < 1% [10]
Figure 1 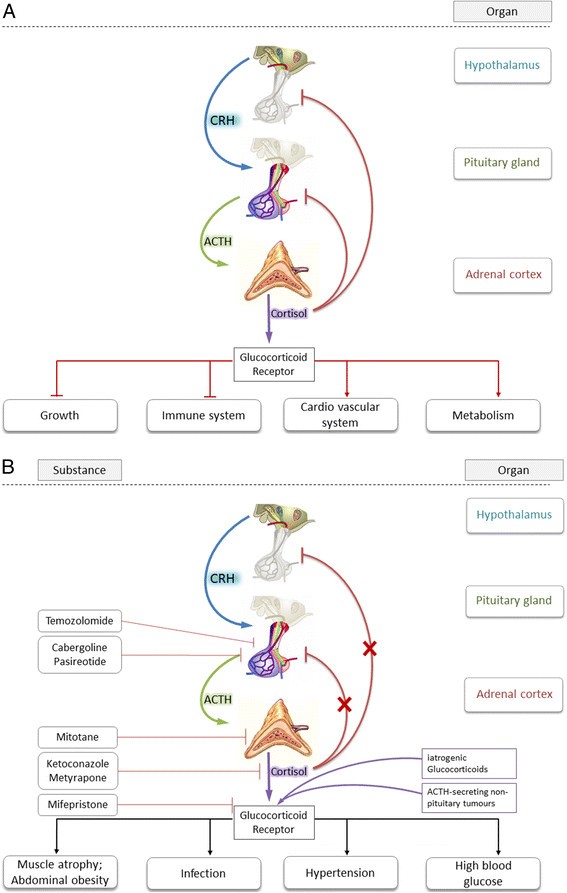
Schematic overview of the hypothalamic-pituitary-adrenal (HPA) axis. A) Schematic overview of the hypothalamic-pituitary-adrenal (HPA) axis. Depicted is the physiological hierarchical pathway to cortisol-secretion from the zona fasciculata of the adrenal cortex including negative feed-back loop. Also shown are the most prominent effects of cortisol. B) Schematic overview of HPA axis in pathologically de-regulated CS-patients. Pathological condition may lead to CRH, ACTH, and cortisol overproduction and impaired negative feed-back loop. Also shown are most hazardous clinical side-effects of hypercortisolism and target of cortisol blockade.

However, systemic treatment options are limited and clinical evidence of these options is scarce (with Pasireotide as the exception from the rule). The regulatory status of pharmacological treatment options are reflected in Table 1. Chemical structures of medicines used in-label and off-label are depicted in Figure 2. According to the European Community Register [11] (accessed on March 12th, 2014) the medicinal products were granted an orphan designation in the context of CS.

Chemical structures of medicinal products used for systemic treatment of CS in alphabetical order.
Etiology and symptoms
CS is a rare disease according to the European orphan regulation [12] affecting not more than 5/10,000 persons in Europe. CS is a heterogeneous disorder that arises from multiple causes and has a broad spectrum of eventually fatal co-morbidities such as diabetes and hypertension.
ERCUSYN is the synonym for The European Registry on Cushing’s Syndrome. In a startling publication from 2011 Valassi et al. [13] describe the baseline demographic and clinical characteristics from:
-
1.
a prospective cohort of 398 CS patients who were recruited from October 1st 2008 (when the database was opened) to October 31st 2010, and
-
2.
a retrospectively collected cohort of 83 patients diagnosed of CS since January 1st 2000 with yearly updates.
This patient population consisted of:
-
➢ 317 (66%) patients suffering from CD
-
➢ 130 (27%) patients who had adrenal-CS
-
➢ 24 (5%) patients who had ectopic-CS
-
➢ 10 (2%) patients classified as having other forms of CS
CS comprises numerous general and endocrine symptoms and side effects some of which might be entailed with fatal outcome. Excess cortisol levels result in (among others):
-
➢ facial plethora
-
➢ hirsutism
-
➢ gonadal dysfunction
-
➢ menstrual irregularities
-
➢ depression
-
➢ infections due to generalized immune suppression
-
➢ striae
-
➢ vascular fragility
-
➢ hypokalemia
-
➢ osteoporosis and eventually fractures
-
➢ muscle weakness
Thus, adverse events are indistinguishable from long-term (sometimes unavoidable) glucocorticoid therapy. The metabolic consequences of cortisol excess include:
-
➢ weight gain
-
➢ central obesity
-
➢ skin atrophy
-
➢ glucose intolerance entailed by diabetes and insulin resistance
-
➢ dyslipidemia
-
➢ hypertension and
-
➢ clotting disorders (eventually even hypercoagulability [14].
Especially weight gain, diabetes, and hypertension are hazardous for patients due to two reasons:
-
1)
They are (apart from infections) responsible for a 50% mortality within the first 5 years after diagnosis mainly due to cardiovascular events [15].
-
2)
These symptoms usually are diagnosed as metabolic syndrome, thereby delaying time to diagnosis [16].
According to current data time to diagnosis after the first symptoms is 6.0 years in mean [17]. In this regard, un-specificity of symptoms is highly problematic alike the slow-progressive nature of the disease [18]. However, months or even years of un-diagnosed CS may cause irreversible organ damage or be fatal due to late-stage diagnosis of malignant disease (i.e. ectopic ACTH producing SCLC). Recovery from the a.m. co-morbidities occurs, but may be delayed or incomplete. The duration of chronic hypercortisolism may determine reversibility of the co-morbidities associated with CD [19]. In addition, CS side-effects lead to an increased cardiovascular risk, thus, rendering even timely diagnosed CS a life-threatening disease, which is also due to infections: 71.4% of the deaths from CS can be attributed to cardiovascular causes or infection [20]. Standardized mortality ratios (SMR) in Cushing patients throughout the Ntali-study were statistically significantly elevated in the overall analysis (SMR 9.3; 95% CI, 6.2 - 13.4, p < 0.001), as well as in all subgroups of patients under investigation. Patients with ectopic CS had the worst outcome with a probability of only 77.6% to survive the next 5 years. A systematic analysis of mortality studies in patients with CS as a consequence of an adrenal adenoma was undertaken by Graversen and colleagues in 2012 [21]. CD patients with persistent disease after initial surgery had a SMR of 3.73 (95% CI: 2.31 - 6.01), whereas mortality of CD patients with initial remission did not differ significantly from the general population (SMR: 1.23 (95% CI: 0.51 - 2.97)). This study confirms the excess mortality in patients with CD if remission after initial surgery is not achieved.
Epidemiology of Cushing’s syndrome as a rare disease
Approximately 1% of the population use exogenous steroids, of which 70% experience one or more adverse events [22]. In contrast, a national register in Denmark reported an annual incidence for endogenous CS of two cases per million people [23]. Endogenous CS is usually (in 70% of cases) a result of a pituitary tumor. In general, CS is rare: the reported incidence of endogenous CS worldwide ranges from 0.7 - 2.4 cases per million per year [24]. CD most commonly affects adults aged 20 - 50 years with a marked female preponderance (1:5 ratio of male vs. female). CS patients are at an overall 4-fold higher mortality rate than age- and gender-matched subjects in the general population [25]. When diagnosed the prevalence of co-morbidities has been reported as follows: 58 – 85% of patients have hypertension, 32 – 41% obesity, 20 – 47% diabetes mellitus, 50 – 81% major depression, 31 – 50% osteoporosis, and 38 – 71% dyslipidemia [19].
Lines of treatment
Depending on the cause of these excessive cortisol concentrations in the patient’s blood, different treatment modalities are possible in routine patient care. Therapy of choice is trans-sphenoidal surgery (TSS) of the pituitary adenoma (in case of ACTH-producing tumors). In patients not amenable for surgery radiotherapy remains an option. Thus, primary lines of therapy in CD (i.e. not medication associated CS) consists of TSS and pituitary irradiation.
TSS is currently the most frequently recommended treatment except for patients who are poor surgical candidates, have invasive tumors, or who refuse surgery. The surgical effectiveness varies depending on expertise in pituitary surgery and the size and extension of the anatomic mass. Hypopituitarism is common after TSS with a range between 13 and 81% [3]. Early remission in terms of cortisol blood level normalization is achieved regularly (Figure 1A). Patients who do not achieve normalization with surgery require additional treatment, usually with radiotherapy and/or medication. If damage to the surrounding normal pituitary tissue occurs during surgery, the patient may require lifelong pituitary hormone replacement. However, an examination of remission and recurrence rates in long-term follow-up studies reveals that potentially up to 40% to 50% of patients could require additional treatment [26]. Thus, systemic (medicinal) treatment of CS may be more important than traditionally thought (Figure 1B).
Pituitary irradiation is usually reserved for patients who have tumor remaining after surgery, for patients who are poor candidates for surgery, and for patients who do not respond adequately to surgery and/or medication. The main disadvantages of radiotherapy are that i) normalization of ACTH secretion may take extended periods of time (eventually years) to occur demanding for medication while success of radiation is awaited, and ii) that patients may develop generalized anterior pituitary insufficiency [27].
Pharmacological therapy applies when these two options are not amenable or refused. In cases when pharmacological therapy becomes necessary, Pasireotide should be used in first-line in ACTH-dependent CS. However, systemic pharmacotherapy of CS often shows – sometimes dose-limiting – side effects. A systematic overview of safety profiles is given in Table 2. The source of data in Table 2 is a systematic recherché in section 4.8 of currently (March 2014) approved SmPCs of licensed medicines. In the following sections drugs used in CS treatment (Figure 2) are referred to in alphabetical order (as in all Tables and Figures). Molecular interaction of the respective drug with cortisol-biosynthesis is depicted in Figure 3, whereas PK parameters of the respective substances are listed in Table 3.

Endogenous biosynthesis and mechanisms of drug inhibition of cortisol.
Aminoglutethimide
Aminoglutethimide from its molecular mechanism is a non-selective, non-steroidal aromatase inhibitor. Due to its non-selective mechanism of action it also inhibits endogenous cortisol synthesis. Schteingart et al. back in 1966 showed that Aminoglutethimide can alleviate clinical symptoms of CS in a patient suffering from metastatic adrenal cancer [37]. However, the same consortium of authors found one year later that its effect can be overruled by excessive secretion of ACTH [38]. Following a report of three patients receiving Aminoglutethimide in combination with Metyrapone, these substances were also given concomitantly [39]. However, Aminoglutethimide is marketed no more.
Cabergoline
Two types of receptors are widely expressed at the surface of pituitary adenoma cells: the somatostatin receptor subtype 5 (SSTR-5) and the dopamine receptor subtype 2 (D2). As mentioned above, SSTR-5 is occupied by Pasireotide prevalently compared to other somatostatins. Dopamine agonists (e.g. bromocriptine, quinagolide, cabergoline) bind to D2 receptors in the pituitary gland.
In CD corticotroph adenomas mainly express D2 receptors (and SSTR-5). Agonists at these receptors inhibit ACTH-release in cell culture of corticotroph adenomas. In those in vitro model systems compounds that target SSTR-5 (like Pasireotide) or D2 (cabergoline) have shown efficacy in subsets of patients in the clinical setting. Combination therapy by administration of both types of compounds yielded promising results.
These clinical reports of enhanced efficacy of combination therapy with SSTR- and dopamine agonist treatment (originally initiated for pituitary adenoma therapy in suppressing growth hormone hypersecretion) lead to the novel concept of somatostatin-dopamine chimeric molecules, e.g. BIM-23A760 [40]. Preliminary in vitro results suggest that the affinity to D2 receptors is crucial for inhibition of prolactin gene expression and growth hormone secretion. However, the impact of these substances on CS in patients remains to be elucidated. In addition, a recent study showed that D2 receptors are also expressed in the majority of ectopic ACTH-producing syndrome (EAS) cases and that cabergoline may decrease cortisol levels in certain subsets of these patients [41].
Etomidate
Etomidate (just like Ketoconazole) from the chemical perspective is an imidazole derivative. It was developed in the sixties as parenteral hypnotic drug [42]. Mechanistically, Etomidate is a centrally acting γ-aminobutyric acid type A (GABA-A) receptor agonist [43]. Low serum cortisol levels resulting in hypoadrenalism was a randomly discovered side effect of Etomidate [44]. Etomidate (like Metyrapone) inhibits the mitochondrial cytochrome P450 (CYP)-dependent adrenal enzyme 11-β-hydroxylase that catalyzes the production of cortisol from deoxycortisol, thereby lowering cortisol blood levels within hours [45]. In a recent systematic review, Preda et al. came to the conclusion that Etomidate might be of clinical benefit in last-line therapy under intensive care condition. In the outpatient endocrinology setting for patients with severe hypercortisolism there might be a role for Etomidate [30]. Etomidate usually is thought to be efficacious only in hypnotic doses, but this appears not to be the case: in the Preda publication, non-hypnotic doses of only 0.3 mg/kg were active in lowering cortisol levels.
Glitazones
Glitazones (Thiazolidinedions) like Rosiglitazone or Pioglitazone are anti-diabetic insulin sensitizing drugs mediating their effects via activation of the transcription factor peroxisome proliferator-activated receptor γ (PPAR γ). Activation of PPARγ results in effects on adipogenesis, carbohydrate and lipid metabolism, inflammation processes, and cell proliferation [46]. Additionally, PPARγ agonists have been shown to inhibit the growth of several tumor cells derived from lung, breast, prostate, and colon cancer tissues [47]–[50]. Immunocytochemical PPARγ expression could be shown in autopsy-derived human pituitary tissue. PPARγ expression was primarily restricted and co-localized with ACTH [51]. However, in pituitary tumors, including ACTH secreting tumors, PPARγ expression is increased as compared to the normal pituitary [52]. In rat and human corticotroph adenoma cell lines Rosiglitazone decreased tumor cell growth, increased apoptosis, and lowered ACTH secretion by inhibiting mRNA expression of its precursor-protein pro-opiomelanocortin (POMC). PPARγ has furthermore been shown to act as tumor suppressor gene in animals [53],[54]. It was hypothesized that glitazones might be useful in treating corticotroph pituitary adenomas by inhibiting ACTH synthesis and secretion and tumor growth. Subsequently, clinical studies investigating the role of glitazones in the treatment of CD were conducted:
In one study, 14 patients with active CD (7 untreated and 7 after unsuccessful surgery) were treated with 8 – 16 mg of Rosiglitazone for 1 – 7 months [55]. In six patients, 24-hour urinary free cortisol (UFC) was significantly lowered and two of them showed clinical improvement at 7-month follow-up. However, after 10 month cortisol levels increased and clinical signs relapsed [56]. Pre-operative Rosiglitazone treatment (8 mg/day) of two patients with pituitary-dependent CS lowered cortisol levels in the 24-hour urine and lead to clinical improvement [57]. In a study of 10 patients, four prior to surgery, four following relapse after surgery, and two immediately after failed surgery treated with 4 – 16 mg of Rosiglitazone for 1 – 8 months, no consistent reductions in UFC levels were found. Only 3 of 10 patients had normalized urinary cortisol levels up to 8 months [58]. Side effects reported included edema, weight gain, somnolence, and increased hirsutism. Although most studies used Rosiglitazone, one study with Pioglitazone is available [59]. In none of the five patients with CD treated with 45 mg Pioglitazone for 30 days any UFC responses were observed. Taken together, glitazone treatment failed to reproduce the effects seen in vitro and in animal studies. Only Rosiglitazone could be shown to be effective at least in a small subset of patients. However, Rosiglitazone is currently suspended in the European Union due to an increased cardiovascular risk.
Ketoconazole
Ketoconazole originally was developed as an anti-fungal medicine. In fact, it was one of the first orally bioavailable systemic anti-mycotic drugs but is also used locally. Chemically it belongs to the first-generation imidazole anti-fungal drugs (in contrast to triazols). Due to its hepatotoxic properties, oral formulations of Ketoconazole are no longer marketed as anti-fungal medicines as recommended by the European Medicines Agency (EMA) after the Committee for Medicinal Products for Human Use (CHMP) concluded that the risk of liver injury is greater than the benefits in treating fungal infections [60]. In fungi it mechanistically acts as an ergosterole synthesis inhibitor by blocking various CYP-dependent enzymes, thereby explaining both, its hepatotoxic side-effects and its efficacy in Cushing’s treatment: Steroidogenesis in the zona fasciculata of the adrenal cortex mainly depends on CYP enzymes [61]. At lower doses, C17-20-lyase and 17-α-hydroxylase are inhibited in both the testicular and adrenal glands. In contrast, cholesterol side-chain cleavage enzymes and 11-β-hydroxylase are inhibited only at higher doses. However, a negative benefit-risk-ratio for one indication (anti-fungal) where alternatives exist does not imperatively mean this also accounts for other indications (Cushing). EMAs safety concern was due to severe hepatic injury but the incidence was only 1 in 15000 cases [62], thus, benefit risk might be different in a disease with high mortality and less alternatives compared to the anti-fungal indication. Currently, Ketoconazole is used off-label in CS, however, marketing authorization applications (MAAs) are underway to change this unsatisfactory status. Thus, future will tell whether Ketoconazole has a positive benefit-risk ratio in CS and currently no final judgment can be made. Ketoconazole may have clinical benefit in CS patients with hyperandrogenic symptoms as it also inhibits steroidogenesis of androgens (for instance in the testes). The occurrence of hirsutism with Metyrapone treatment in women may lead a clinician to choose Ketoconazole in this case or may lead to a combination therapy of Metyrapone and Ketoconazole. Some patients even may require combination of both to achieve control of CS. The dose of Ketoconazole used as monotherapy in patients with CD ranges from 200 mg to 1200 mg per day [63].
Metyrapone
Metyrapone (Metopirone®) is only registered in four European countries (France, United Kingdom, the Netherlands, and Ireland) through national procedures. From the chemical structure, Metyrapone is a very simple molecule displaying no stereochemistry (2-methyl-1,2-di(pyridin-3-yl)propan-1-one). The drug is very old as it was introduced into the pharmacological armamentarium in the late 50ties of the last century. Thus, in most countries, it was not licensed in a modern way based on careful assessment of quality, safety, and efficacy, but registered and this is the reason why evidence whether Metyrapone is a worthy option in the treatment of CS is rather low. Most publications on the clinical use of Metyrapone are case studies or case series, eventually very small, non-controlled studies. Thus, licensing status is very different among ICH (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use) countries. Metyrapone is more frequently used in the UK in Ireland, while it is not even licensed in Germany, Italy or Spain (to name just a few). Metyrapone is an inhibitor of 11-β-hydroxylase thereby inhibiting endogenous cortisol synthesis. In return, ACTH synthesis is triggered, and this forms the basis for Metyrapone functional test in diagnosis of a properly functioning hypothalamic-pituitary-adrenal (HPA) axis. However, the substance is not listed in current diagnosis guidelines for CS. According to current medicinal guidelines, diagnosis of hypercortisolism (irrespective of its origin) is performed according to the following scheme:
-
1.
Complete blood count including serum electrolytes, blood sugar etc.
-
2.
UFC from 24 h-urine sampling and circadian profile of plasma cortisol, plasma ACTH, dehydroepiandrosterone, testosterone itself, and urine steroid profile
-
3.
Low-Dose-Dexamethasone-Test, High-Dose-Dexamethasone-Test
-
4.
After endocrine diagnostic tests: magnetic resonance imaging (MRI), ultra-sound, computer tomography (CT) and other localization diagnostics
This diagnosis flow-scheme also is reflected in current international text books [64] and for instance in the clinical practice guidelines of the endocrine society [65]:
“After excluding exogenous glucocorticoid use, we recommend testing for Cushing’s syndrome in patients with multiple and progressive features compatible with the syndrome, particularly those with a high discriminatory value, and patients with adrenal incidentaloma. We recommend initial use of one test with high diagnostic accuracy (urine cortisol, late night salivary cortisol, 1 mg overnight or 2 mg 48-h dexamethasone suppression test). We recommend that patients with an abnormal result see an endocrinologist and undergo a second test, either one of the above or, in some cases, a serum midnight cortisol or dexamethasone-CRH test.”
Metyrapone, thus, is an excellent example for national traditions determining clinical management of rare diseases. In CS treatment usually is a case-by-case decision because of the heterogeneity of the patients’ population especially in regard to their co-morbidities. Consequently, no (national or international) medicinal guidelines or treatment algorithms of high evidence for management of CS exist. A consensus statement on ACTH-dependent CS published in 2008 by Biller et al. emphasized clinical management of CS as a multi-disciplinary approach on an individualized basis [66].
Despite its efficacy in lowering cortisol blood levels, Metyrapone is not constantly effective and therefore does not cover the entire CS-population. Clinical and/or biochemical improvements can be achieved in 60 to 80% of patients and are usually associated with improvements in symptoms. In general, Metyrapone appeared to be well tolerated, but may sometimes lead to an accumulation of androgens in women during long-term treatment (hirsutism).
Mifepristone
Mifepristone (experimental name RU-486) originally was developed as the so-called “abortion pill”. Chemically it is an all-trans steroid substituted at positions 11 and 17. Mechanistically, it is an antagonist at the progesterone receptor (PR), thereby explaining its acute abortive effect. It also shows antagonistic activity at the glucocorticoid receptor (GR) with a higher affinity than Dexamethasone. Thus, unlike other agents, Mifepristone does not decrease cortisol synthesis but directly antagonizes its effects. Therefore, it is intuitive that patients dosed with Mifepristone can hardly be diagnosed by serum cortisol measurements as they remain unchanged. Mifepristone is not approved in the indication CS. Side effects include decreased plasma potassium levels with a potential for heart conduction abnormalities, vaginal bleeding, and endometrial thickening (not surprising for a progesterone antagonist).
Notions on Mifepristone
Apart from the fact that everyone knows it as an abortion pill, it must not be forgotten that Mifepristone originally was developed as an anti-glucocorticoid. Soon afterwards, it was found to block PRs intrinsic activity. Due to its pronounced cortisol-blocking effect, new generation antiprogestins were developed aiming at reducing anti-glucocorticoid activity while maintaining anti-progesterone activity (i.e. ORG-31710, CDB-2914, CDB-4124). However, only Mifepristone has been licensed by US Food and Drug Administration (FDA) to terminate early pregnancy (working as an antiprogestin) or ameliorating the hyperglycemia in CS-patients [67]. In contrast, other PR antagonists like ulipristal and proellex are currently investigated for their potential to alleviate symptoms in endometriosis and uterine fibroids, thereby approaching indications like other hormone ablative medicines (i.e. GnRH analogues) [68].
Interestingly, Tieszen et al. investigated the potential of PR antagonists, to inhibit the growth of cells from endocrine related cancers (i.e. ovarian-, breast-, and prostate cancer cells), expressing different sets of hormone receptors. The authors found, that all cancer cells were inhibited in growth irrespective of their PR status, as there are: MCF-7 breast cancer cells carrying PR, MDA-MB- 231 breast cancer cells with no PR expression, PR negative and androgen receptor positive LNCaP prostate cancer cells, and PR negative androgen receptor positive PC3 prostate cancer cells are all inhibited by mifepristone with similar potency [69].
However, all cell lines under investigation in this study express GR and derivatives of mifepristone with lower GR affinity are less effective. Therefore, it can be concluded that GR action might contribute to anti-tumor effects of PR antagonists. Thus, PRs may not be required for the inhibition of cancer growth triggered by Mifepristone. In addition, it has been shown by another group, that Mifepristone blocked the growth of estrogen receptor negative and PR negative MDA-MB-231 breast cancer cells [70].
Mitotane
Mitotane is another unusual drug used in certain very rare cases of CS. Mitotane (Lysodren®) is authorized only for symptomatic treatment of advanced (very rare) adrenal cortical carcinoma (ACC). Chemically it is an ortho-derivative of the long-term known insecticide DDT (dichloro-diphenyl-trichloroethane), the bis-trans-form of the molecule. Its biochemical mechanism is not yet fully understood, but inhibition of side-chain cleavage of cholesterol (a decisive step in endogenous steroidogenesis) seems to play a role as well as blockade of 3-β-hydroxysteroid-deshydrogenase [71]. Like its mother substance DDT Mitotane also accumulates in adipose tissue, which might then be entailed with longer half-life. Especially in Cushing’s patients this is of particular concern, as these patients usually present with higher fat mass compared to age-adjusted matched population. Therefore, it usually takes weeks until complete efficacy is reached. In the current SmPC it is even mentioned that 3 - 5 months can be assumed until the intended plasma level of 14 - 20 mg/L is reached. This window should be reached (titrated) by monitoring patients’ blood levels. Thus, Mitotane fulfills the requirements of a Narrow Therapeutic Index Drug (NTID) according to the FDA definition. Mitotane has an orphan designation and, thus, the pivotal registration trial was performed in 177 patients only showing an increase in recurrence-free interval after radical surgery followed by Mitotane compared to surgery alone [72]. Grade 3 side effects were mainly neurologic (confusion, ataxia, vertigo) or biochemical (elevated γ-glutamyltransferase). Thus, Mitotane might be of clinical benefit in some patients, adjuvant to radical surgery.
Pasireotide and somatostatin analogues
Pasireotide mechanistically is a somatostatin-analogue (SSA). SSAs are the first-line medical treatment in GH-secreting adenomas (with the clinical manifestation of acromegaly). Currently two different molecular entities, octreotide (Sandostatin®) and lanreotide (Somatuline®), are marketed. Octreotide, which was licensed 25 years ago, is available as a short-acting subcutaneous formulation for twice-daily administration, and a long-acting (LAR) microsphere preparation administered by intra muscular injection every 4 weeks. Lanreotide is available in a microsphere formulation (sustained release) and a high-concentrate aqueous solution (Autogel). From clinical safety point of view SSA side effects include gallstones and gastrointestinal affection (diarrhea, nausea, abdominal pain). According to the route of administration injection site reactions (pain, swelling) have also been reported.
Pasireotide (experimental name SOM230) itself is a cyclic hexapeptide and was licensed via the central route in Europe in 2012 as first-line pharmacological therapy option. The European Public Assessment Report (EPAR) is dated June 1st 2012 [73].
Endogenous somatostatin is a peptide hormone widely distributed in the endocrine system. Somatostatin action is mediated through five different SSTR subtypes (SSTR-1-SSTR-5). SSTRs belong to the superfamily of tripartite G-protein coupled receptors. They are expressed over the whole body in various tissues, with cells from different tissues expressing different receptor subtypes at different densities. The physiological actions of somatostatin are numerous. It is an inhibitory protein of endocrine secretion of various organs, including the pituitary, pancreas, gastrointestinal tract, thyroid, kidney, and adrenal glands. Among others, it inhibits gallbladder contractility and bile flow, and stimulates gastrointestinal water and electrolyte absorption. Pasireotide has a slightly different SSTR binding profile than the first-generation SSAs Octreotide and Lanreotide, with high affinity to four of the five receptors (SSTR-1, −2, −3 and −5). Compared to Octreotide the binding affinity of Pasireotide is 30 - 40 times greater for SSTR-1 and SSTR-5 and 5 times greater for SSTR-3, whereas the affinity for SSTR-2 is comparable.
In general, medicines for the treatment of CS (i.e. Mifepristone, Ketoconazole) are not selective and initially were not developed for the treatment of CS. In contrast, Pasireotide exhibits at least a subtype-prevalence for subtype-5 of the SSTR in the pituitary gland. SSTR-5 prevalently is expressed at the surface of ACTH secreting cells, thus showing at least partial specificity in lowering excessive cortisol blood levels. Therefore, first-line pharmacological therapy is Pasireotide injection (usually while definitive therapy is awaited).
In addition, the basis of the centralized approval for Pasireotide was a clinical development program consisting (only regarding patients) of
-
1.
a 22-patient phase II study reviewed in [74]
-
2.
a randomized-dose, double-blinded pivotal phase III study [75].
The study design was as follows:
162 patients with Cushing’s disease were randomized (double-blind) to pasireotide 600 μg (n = 82) or 900 μg (n = 80) sc bid. After 3mo, patients with UFC > 2 × ULN (ULN: 145 nmol/24 h) or UFC > baseline were unblinded and the dose increased by 300 μg bid. All others continued on the same double-blind dose to 6mo. Months 6–12 were open-label with dose titration performed when needed. Primary endpoint: UFC ≤ ULN at 6mo without dose up-titration from the randomized dose.
However, it should be noticed that Pasireotide is indicated only in a subgroup of CD-patients and shows satisfactory efficacy only in ~ 30% of patients. Side effects with this agent are similar to those of other SSAs. Hyperglycemia-associated side-effects are of particular concern in CS patients and were reported in 73% of patients in the pivotal study.
Temozolomide
Temozolomide (Temodal®) is licensed for Grade III and IV glioblastoma multiforme concomitant to radiotherapy [76]. Temozolomide is an orally bioavailable, centrally active alkylating agent. Following a systematic review from 2012 up to now, 46 cases of adenohypophysial tumors are published in Medline that were treated with Temozolomide (30 adenomas, 16 carcinomas). 60% of the adenomas and 69% of carcinomas responded favorably to treatment [77]. Thus, in (rare) cases of highly aggressive growing macroademomas or pituitary carcinomas Temozolomide might be an option.
Trilostane (only in veterinary use)
Chemically Trilostane is a 2-cyano-4,5-epoxy-steroid. Trilostane reversibly suppresses adrenal function. From its molecular mechanism of action it inhibits cortisol biosynthesis by prevalently blocking 3-β-hydroxysteroide-dehydrogenase. This, however, does not only block cortisol- and corticosteron-synthesis, but also biosynthesis of aldosterone. Thus, Trilostane un-specifically blocks both, glucocorticoids and mineralocorticoids. High doses are reported to even block gonadal steroidogenesis. However, Trilostane is not used as human medicine; its only indication is CS in dogs, irrespective whether CS derives from pituitary or adrenal cortisol-overproduction. For many years before Mitotane has been considered the medical treatment of choice for dogs with adrenal-dependent hyperadrenocorticism [78].
Therapy of Cushing’s syndrome in children
First of all, in the pediatric population no final conclusion can be drawn on any of the medicines under investigation due to lack of pivotal evidence. This is best reflected by the fact that the EMA accepted a waiver for clinical data from pediatric patients for centrally approved Pasireotide (a so-called “PIP-Waiver”; PIP, pediatric investigation plan). Throughout the SmPCs it is mentioned that only limited evidence is available for children and, thus, the respective medicine is not recommended for use in children. However, in those drugs used off-label even this is based on other indications. There is only one exemption from the rule; for Metyrapone the following can be read in the SmPC: “In children the dosage should be 15 mg/kg bodyweight, with a minimum dose of 250 mg every 4 hours for 6 doses.” However, even for Metyrapone evidence in children is very low (15 documented cases) [2],[79],[80].
Co-medication for symptomatic therapy
Clinical management of co-morbidities of CS is a complex challenge. Post-surgery management of CS patients is tri-phasic. Ragnarsson and Johannsson summarized systemic therapy while awaiting success of surgery (or radiotherapy) [3].
Usually supportive therapy of CS co-morbidities is by individual therapy depending on clinical presentation of the patient. Certain hazardous (life-threatening) symptoms are treated on a case-by-case decision according to medicinal guidelines of the respective symptoms. The following medicinal guidelines may serve as examples for clinical management of most hazardous co-morbidities:
Conclusion
CS can arise from different pathological conditions ranging from benign pituitary adenomas to adrenal carcinomas or ectopic cortisol secretion. Thus, various drugs with different target structures and mechanisms of action are used in systemic therapy of CS comprising somatostatin-analogues, cortisol synthesis inhibitors, receptor antagonists and unusual substances like Temozolomide and Mitotane for certain very rare conditions. Some mechanisms of action are well known (Pasireotide, Ketoconazole) others are not fully understood (Mitotane).These drugs differ in terms of safety profile, route of administration, and indications considerably. In addition, safety profiles differ considerably and become hardly predictable, if combinations are needed due to insufficient clinical control of CS by only one substance. Many of these substances are used off-label and constitute an ultima ratio approach after failure of preceding therapy options. Thus, clinical and/or at least epidemiological data will be needed to finally judge on safety and efficacy in regard of a debilitating underlying disease.
Search strategy
Recherché of clinical PK-parameter and special populations
i) SmPCs as published on the heads of medicines agencies (HMA) homepage (http://mri.medagencies.org/Human/) were accessed in March and June 2014. Section 5.2 (Pharmacokinetics) of SmPCs systematically was searched for PK-parameters.
ii) A search in PubMed/Medline was performed in March and June 2014 using the international non-proprietary name (INN) of the respective medicinal product combined with the terms “cushing” and “indication” as well as “cushing” and “pediatric”. PubMed recherché was restricted by searching only in clinical trials.
Safety profile assessment
SmPCs of currently marketed medicinal products were accessed (if not licensed for treatment of CS, then in other indication; i.e. Ketoconazole, Etomidate, Temozolomide, and Pioglitazone). If not marketed any more (Aminoglutethimide) the last approved SmPC was used except for Trilostane, were veterinary SmPC of Swissmedic was used. Side-effects were categorized by frequency for preferred terms (PT) of the systems organ class (SOC) system (XX = common, very common; X = rare, uncommon; not known).
Abbreviations
- ACC:
-
Adrenal cortical carcinoma
- ACTH:
-
Adreoncorticotropic hormone
- AUC:
-
Area under the curve
- b.i.d.:
-
Twice daily
- CD:
-
Cushing’s disease
- CMR:
-
Carcinogenic, mutagenic and toxic for reproductive system
- CS:
-
Cushing’s syndrome
- CT:
-
Computer tomography
- CHMP:
-
Committee for medicinal products for human use
- Cmax:
-
Maximal plasma concentration
- CRH:
-
Corticotropin-releasing hormone
- CYP:
-
Cytochrome P450
- D2:
-
Dopamine receptor subtype 2
- EAS:
-
Ectopic ACTH-producing syndrome
- EMA:
-
European medicines agency
- EPAR:
-
European public assessment report
- FDA:
-
US food and drug administration
- GABA-A:
-
γ-aminobutyric acid type A
- GR:
-
Glucocorticoid receptor
- HMA:
-
Heads of medicines agencies
- HPA:
-
Hypothalamic-pituitary-adrenal axis
- ICH:
-
International conference on harmonisation of technical requirements for registration of pharmaceuticals for human use
- INN:
-
Non-proprietary name
- MAA:
-
Marketing authorization application
- MRI:
-
Magnetic resonance imaging
- NTID:
-
Narrow therapeutic index drug
- PIP:
-
Pediatric investigation plan
- POMC:
-
Pro-opiomelanocortin
- PK:
-
Pharmacokinetics
- PR:
-
Progesterone receptor
- PT:
-
Preferred terms
- q.d.:
-
Every day
- SCLC:
-
Small-cell lung cancer
- SmPC:
-
Summary of product characteristics
- SMR:
-
Standardized mortality ratios
- SOC:
-
Systems organ class
- SSA:
-
Somatostatin-analogue
- SSTR:
-
Somatostatine receptor
- tmax:
-
Time after administration when Cmax is reached
- TSS:
-
Trans-sphenoidal surgery
- UFC:
-
Urinary free cortisol
References
Purnell JQ, Brandon DD, Isabelle LM, Loriaux DL, Samuels MH: Association of 24-hour cortisol production rates, cortisol-binding globulin, and plasma-free cortisol levels with body composition, leptin levels, and aging in adult men and women. J Clin Endocrinol Metab. 2004, 89: 281-287. 10.1210/jc.2003-030440.
Jackman S, Diamond F: Pituitary adenomas in childhood and adolescence. Pediatr Endocrinol Rev. 2013, 10: 450-459.
Ragnarsson O, Johannsson G: Cushing’s syndrome: a structured short- and long-term management plan for patients in remission. Eur J Endocrinol. 2013, 169: R139-R152. 10.1530/EJE-13-0534.
Mazziotti G, Giustina A: Glucocorticoids and the regulation of growth hormone secretion. Nat Rev Endocrinol. 2013, 9: 265-276.
Lake MG, Krook LS, Cruz SV: Pituitary adenomas: an overview. Am Fam Physician. 2013, 88: 319-327.
Gustafsson BI, Kidd M, Chan A, Malfertheiner MV, Modlin IM: Bronchopulmonary neuroendocrine tumors. Cancer. 2008, 113: 5-21. 10.1002/cncr.23542.
Shahani S, Nudelman RJ, Nalini R, Kim HS, Samson SL: Ectopic corticotropin-releasing hormone (CRH) syndrome from metastatic small cell carcinoma: a case report and review of the literature. Diagn Pathol. 2010, 5: 56-10.1186/1746-1596-5-56.
Bourdeau I, Lampron A, Costa MH, Tadjine M, Lacroix A: Adrenocorticotropic hormone-independent Cushing’s syndrome. Curr Opin Endocrinol Diabetes Obes. 2007, 14: 219-225. 10.1097/MED.0b013e32814db842.
Courcoutsakis N, Prassopoulos P, Stratakis CA: CT findings of primary pigmented nodular adrenocortical disease: rare cause of ACTH-independent Cushing syndrome. AJR Am J Roentgenol. 2010, 194: W541-10.2214/AJR.09.4056.
Maghrabi A, Yaqub A, Denning KL, Benhamed N, Faiz S, Saleem T: Challenges in the diagnostic work-up and management of patients with subclinical Cushing’s syndrome and bilateral adrenal masses. Endocr Pract. 2013, 19: 515-521. 10.4158/EP12277.RA.
European Commission, Communitiy register of medicinal products. , : [http://ec.europa.eu/health/documents/community-register/]
Regulation (EC) No 141/2000 of the European Parliament and of the Council on Orphan Medicinal Products. ,:, [http://eur-lex.europa.eu/]
Valassi E, Santos A, Yaneva M, Toth M, Strasburger CJ, Chanson P, Wass JA, Chabre O, Pfeifer M, Feelders RA, Tsagarakis S, Trainer PJ, Franz H, Zopf K, Zacharieva S, Lamberts SW, Tabarin A, Webb SM: The European Registry on Cushing’s syndrome: 2-year experience. Baseline demographic and clinical characteristics. Eur J Endocrinol. 2011, 165: 383-392. 10.1530/EJE-11-0272.
van der Pas R, Leebeek FW, Hofland LJ, de Herder WW, Feelders RA: Hypercoagulability in Cushing’s syndrome: prevalence, pathogenesis and treatment. Clin Endocrinol (Oxf). 2013, 78: 481-488. 10.1111/cen.12094.
Plotz CM, Knowlton AI, Ragan C: The natural history of Cushing’s syndrome. Am J Med. 1952, 13: 597-614. 10.1016/0002-9343(52)90027-2.
Prague JK, May S, Whitelaw BC: Cushing’s syndrome. BMJ. 2013, 346: f945-10.1136/bmj.f945.
Psaras T, Milian M, Hattermann V, Freiman T, Gallwitz B, Honegger J: Demographic factors and the presence of comorbidities do not promote early detection of Cushing’s disease and acromegaly. Exp Clin Endocrinol Diabetes. 2011, 119: 21-25. 10.1055/s-0030-1263104.
Ross EJ, Linch DC: Cushing’s syndrome–killing disease: discriminatory value of signs and symptoms aiding early diagnosis. Lancet. 1982, 2: 646-649. 10.1016/S0140-6736(82)92749-0.
Feelders RA, Pulgar SJ, Kempel A, Pereira AM: The burden of Cushing’s disease: clinical and health-related quality of life aspects. Eur J Endocrinol. 2012, 167: 311-326. 10.1530/EJE-11-1095.
Ntali G, Asimakopoulou A, Siamatras T, Komninos J, Vassiliadi D, Tzanela M, Tsagarakis S, Grossman AB, Wass JA, Karavitaki N: Mortality in Cushing’s syndrome: systematic analysis of a large series with prolonged follow-up. Eur J Endocrinol. 2013, 169: 715-723. 10.1530/EJE-13-0569.
Graversen D, Vestergaard P, Stochholm K, Gravholt CH, Jorgensen JO: Mortality in Cushing’s syndrome: a systematic review and meta-analysis. Eur J Intern Med. 2012, 23: 278-282. 10.1016/j.ejim.2011.10.013.
Fardet L, Flahault A, Kettaneh A, Tiev KP, Genereau T, Toledano C, Lebbé C, Cabane J: Corticosteroid-induced clinical adverse events: frequency, risk factors and patient’s opinion. Br J Dermatol. 2007, 157: 142-148. 10.1111/j.1365-2133.2007.07950.x.
Lindholm J, Juul S, Jorgensen JO, Astrup J, Bjerre P, Feldt-Rasmussen U, Hagen C, Jørgensen J, Kosteljanetz M, Kristensen L, Laurberg P, Schmidt K, Weeke J: Incidence and late prognosis of cushing’s syndrome: a population-based study. J Clin Endocrinol Metab. 2001, 86: 117-123.
Newell-Price J, Bertagna X, Grossman AB, Nieman LK: Cushing’s syndrome. Lancet. 2006, 367: 1605-1617. 10.1016/S0140-6736(06)68699-6.
Etxabe J, Vazquez JA: Morbidity and mortality in Cushing’s disease: an epidemiological approach. Clin Endocrinol (Oxf). 1994, 40: 479-484. 10.1111/j.1365-2265.1994.tb02486.x.
Fleseriu M: Medical management of persistent and recurrent cushing disease. Neurosurg Clin N Am. 2012, 23: 653-668. 10.1016/j.nec.2012.06.012.
Bodaghabadi M, Riazi H, Aran S, Bitaraf MA, Alikhani M, Alahverdi M, Mohamadi M, Shalileh K, Azar M: Repeated transsphenoidal surgery or gamma knife radiosurgery in recurrent cushing disease after transsphenoidal surgery. J Neurol Surg A Cent Eur Neurosurg. 2014, 75: 91-97.
Del DP, Bonuccelli U: Clinical pharmacokinetics of cabergoline. Clin Pharmacokinet. 2003, 42: 633-645. 10.2165/00003088-200342070-00003.
Allolio B, Schulte HM, Kaulen D, Reincke M, Jaursch-Hancke C, Winkelmann W: Nonhypnotic low-dose etomidate for rapid correction of hypercortisolaemia in Cushing’s syndrome. Klin Wochenschr. 1988, 66: 361-364. 10.1007/BF01735795.
Preda VA, Sen J, Karavitaki N, Grossman AB: Etomidate in the management of hypercortisolaemia in Cushing’s syndrome: a review. Eur J Endocrinol. 2012, 167: 137-143. 10.1530/EJE-12-0746.
European Public Assessment Report - Scientific Discussion of Lysodren. 2005, EMEA/H/C/000521, London
Brada M, Judson I, Beale P, Moore S, Reidenberg P, Statkevich P, Dugan M, Batra V, Cutler D: Phase I dose-escalation and pharmacokinetic study of temozolomide (SCH 52365) for refractory or relapsing malignancies. Br J Cancer. 1999, 81: 1022-1030. 10.1038/sj.bjc.6690802.
Liu JH, Garzo VG, Yen SS: Pharmacodynamics of the antiprogesterone RU486 in women after oral administration. Fertil Steril. 1988, 50: 245-249.
Castinetti F, Morange I, Conte-Devolx B, Brue T: Cushing’s disease. Orphanet J Rare Dis. 2012, 7: 41-10.1186/1750-1172-7-41.
Heikinheimo O: Pharmacokinetics of the antiprogesterone RU 486 in women during multiple dose administration. J Steroid Biochem. 1989, 32: 21-25. 10.1016/0022-4731(89)90008-3.
Beglinger C, Hu K, Wang Y, Bouillaud E, Darstein C, Wang Y, Mohideen P: Multiple once-daily subcutaneous doses of pasireotide were well tolerated in healthy male volunteers: a randomized, double-blind, placebo-controlled, cross-over, Phase I study. Endocrine. 2012, 42: 366-374. 10.1007/s12020-012-9668-1.
Schteingart DE, Cash R, Conn JW: Amino-glutethimide and metastatic adrenal cancer. Maintained reversal (six months) of Cushing’s syndrome. JAMA. 1966, 198: 1007-1010. 10.1001/jama.1966.03110220091030.
Schteingart DE, Conn JW: Effects of aminoglutethimide upon adrenal function and cortisol metabolism in Cushing’s syndrome. J Clin Endocrinol Metab. 1967, 27: 1657-1666. 10.1210/jcem-27-12-1657.
Thoren M, Adamson U, Sjoberg HE: Aminoglutethimide and metyrapone in the management of Cushing’s syndrome. Acta Endocrinol (Copenh). 1985, 109: 451-457.
Culler MD: Somatostatin-dopamine chimeras: a novel approach to treatment of neuroendocrine tumors. Horm Metab Res. 2011, 43: 854-857. 10.1055/s-0031-1287769.
de Bruin C, Feelders RA, Lamberts SW, Hofland LJ: Somatostatin and dopamine receptors as targets for medical treatment of Cushing’s syndrome. Rev Endocr Metab Disord. 2009, 10: 91-102. 10.1007/s11154-008-9082-4.
Gooding JM, Corssen G: Etomidate: an ultrashort-acting nonbarbiturate agent for anesthesia induction. Anesth Analg. 1976, 55: 286-289.
Forman SA: Clinical and molecular pharmacology of etomidate. Anesthesiology. 2011, 114: 695-707. 10.1097/ALN.0b013e3181ff72b5.
Watt I, Ledingham IM: Mortality amongst multiple trauma patients admitted to an intensive therapy unit. Anaesthesia. 1984, 39: 973-981. 10.1111/j.1365-2044.1984.tb08885.x.
Wagner RL, White PF, Kan PB, Rosenthal MH, Feldman D: Inhibition of adrenal steroidogenesis by the anesthetic etomidate. N Engl J Med. 1984, 310: 1415-1421. 10.1056/NEJM198405313102202.
Issemann I, Green S: Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990, 347: 645-650. 10.1038/347645a0.
Yang Z, Bagheri-Yarmand R, Balasenthil S, Hortobagyi G, Sahin AA, Barnes CJ, Kumar R: HER2 regulation of peroxisome proliferator-activated receptor gamma (PPARgamma) expression and sensitivity of breast cancer cells to PPARgamma ligand therapy. Clin Cancer Res. 2003, 9: 3198-3203.
Yamada R, Sano H, Hla T, Hashiramoto A, Kawahito Y, Mukai S, Kohno M, Tsubouchi Y, Inoue M, Komatsu A, Inoue K, Kondo M: Selective inhibition of cyclooxygenase-2 with antisense oligodeoxynucleotide restricts induction of rat adjuvant-induced arthritis. Biochem Biophys Res Commun. 2000, 269: 415-421. 10.1006/bbrc.2000.2303.
Nagata D, Yoshihiro H, Nakanishi M, Naruyama H, Okada S, Ando R, Tozawa K, Kohri K: Peroxisome proliferator-activated receptor-gamma and growth inhibition by its ligands in prostate cancer. Cancer Detect Prev. 2008, 32: 259-266. 10.1016/j.cdp.2008.05.008.
Voutsadakis IA: Peroxisome proliferator-activated receptor gamma (PPARgamma) and colorectal carcinogenesis. J Cancer Res Clin Oncol. 2007, 133: 917-928.
Heaney AP, Fernando M, Yong WH, Melmed S: Functional PPAR-gamma receptor is a novel therapeutic target for ACTH-secreting pituitary adenomas. Nat Med. 2002, 8: 1281-1287. 10.1038/nm784.
Heaney AP, Fernando M, Melmed S: Functional role of estrogen in pituitary tumor pathogenesis. J Clin Invest. 2002, 109: 277-283. 10.1172/JCI0214264.
Gruszka A, Kunert-Radek J, Pawlikowski M: Rosiglitazone, PPAR-gamma receptor ligand, decreases the viability of rat prolactin-secreting pituitary tumor cells in vitro. Neuro Endocrinol Lett. 2005, 26: 51-54.
Bogazzi F, Ultimieri F, Raggi F, Russo D, Vanacore R, Guida C, Viacava P, Cecchetti D, Acerbi G, Brogioni S, Cosci C, Gasperi M, Bartalena L, Martino E: PPARgamma inhibits GH synthesis and secretion and increases apoptosis of pituitary GH-secreting adenomas. Eur J Endocrinol. 2004, 150: 863-875. 10.1530/eje.0.1500863.
Ambrosi B, Dall’Asta C, Cannavo S, Libe R, Vigo T, Epaminonda P, Chiodini I, Ferrero S, Trimarchi F, Arosio M, Beck-Peccoz P: Effects of chronic administration of PPAR-gamma ligand rosiglitazone in Cushing’s disease. Eur J Endocrinol. 2004, 151: 173-178. 10.1530/eje.0.1510173.
Cannavo S, Arosio M, Almoto B, Dall’Asta C, Ambrosi B: Effectiveness of long-term rosiglitazone administration in patients with Cushing’s disease. Clin Endocrinol (Oxf). 2005, 63: 118-119. 10.1111/j.1365-2265.2005.02285.x.
Hull SS, Sheridan B, Atkinson AB: Pre-operative medical therapy with rosiglitazone in two patients with newly diagnosed pituitary-dependent Cushing’s syndrome. Clin Endocrinol (Oxf). 2005, 62: 259-261. 10.1111/j.1365-2265.2005.02193.x.
Pecori GF, Scaroni C, Arvat E, Martin M, Giordano R, Albiger N, Leao AA, Picu A, Mantero F, Cavagnini F: Effect of protracted treatment with rosiglitazone, a PPARgamma agonist, in patients with Cushing’s disease. Clin Endocrinol (Oxf). 2006, 64: 219-224. 10.1111/j.1365-2265.2006.02452.x.
Suri D, Weiss RE: Effect of pioglitazone on adrenocorticotropic hormone and cortisol secretion in Cushing’s disease. J Clin Endocrinol Metab. 2005, 90: 1340-1346. 10.1210/jc.2004-1746.
European Medicines Agency recommends suspension of marketing authorisations for oral ketoconazole. 2013, EMA/458028/2013, London
Loose DS, Kan PB, Hirst MA, Marcus RA, Feldman D: Ketoconazole blocks adrenal steroidogenesis by inhibiting cytochrome P450-dependent enzymes. J Clin Invest. 1983, 71: 1495-1499. 10.1172/JCI110903.
Castinetti F, Guignat L, Giraud P, Muller M, Kamenicky P, Drui D, Caron P, Luca F, Donadille B, Vantyghem MC, Bihan H, Delemer B, Raverot G, Motte E, Philippon M, Morange I, Conte-Devolx B, Quinquis L, Martinie M, Vezzosi D, Le Bras M, Baudry C, Christin-Maitre S, Goichot B, Chanson P, Young J, Chabre O, Tabarin A, Bertherat J, Brue T: Ketoconazole in Cushing’s disease: is it worth a try?. J Clin Endocrinol Metab. 2014, 99 (5): 1623-30. 10.1210/jc.2013-3628.
Castinetti F, Morange I, Jaquet P, Conte-Devolx B, Brue T: Ketoconazole revisited: a preoperative or postoperative treatment in Cushing’s disease. Eur J Endocrinol. 2008, 158: 91-99. 10.1530/EJE-07-0514.
Fauci AS, Longo DL, Hauser SL, Jameson JL, Loscalzo J, Kasper DL: Disorders of the Anterior Pituitary and Hypothalamus. Harrison’s Principles of Internal Medicine: vol 18. 2011, Chapter 339, Page 2897-2899; 2011; ISBN 978-0-07174889-6
Nieman LK, Biller BM, Findling JW, Newell-Price J, Savage MO, Stewart P, Montori VM: The diagnosis of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008, 93: 1526-1540. 10.1210/jc.2008-0125.
Biller BM, Grossman AB, Stewart PM, Melmed S, Bertagna X, Bertherat J, Buchfelder M, Colao A, Hermus AR, Hofland LJ, Klibanski A, Lacroix A, Lindsay JR, Newell-Price J, Nieman LK, Petersenn S, Sonino N, Stalla GK, Swearingen B, Vance ML, Wass JA, Boscaro M: Treatment of adrenocorticotropin-dependent Cushing’s syndrome: a consensus statement. J Clin Endocrinol Metab. 2008, 93: 2454-2462. 10.1210/jc.2007-2734.
Telleria CM: Drug repurposing for cancer therapy. J Cancer Sci Ther. 2012, 4: ix-xi. 10.4172/1948-5956.1000e108.
Spitz IM: Clinical utility of progesterone receptor modulators and their effect on the endometrium. Curr Opin Obstet Gynecol. 2009, 21: 318-324. 10.1097/GCO.0b013e32832e07e8.
Tieszen CR, Goyeneche AA, Brandhagen BN, Ortbahn CT, Telleria CM: Antiprogestin mifepristone inhibits the growth of cancer cells of reproductive and non-reproductive origin regardless of progesterone receptor expression. BMC Cancer. 2011, 11: 207-10.1186/1471-2407-11-207.
Liang Y, Hou M, Kallab AM, Barrett JT, El EF, Schoenlein PV: Induction of antiproliferation and apoptosis in estrogen receptor negative MDA-231 human breast cancer cells by mifepristone and 4-hydroxytamoxifen combination therapy: a role for TGFbeta1. Int J Oncol. 2003, 23: 369-380.
Martz F, Straw JA: The in vitro metabolism of 1-(o-chlorophenyl)-1-(p-chlorophenyl)-2,2-dichloroethane (o, p’-DDD) by dog adrenal mitochondria and metabolite covalent binding to mitochondrial macromolecules: a possible mechanism for the adrenocorticolytic effect. Drug Metab Dispos. 1977, 5: 482-486.
Terzolo M, Angeli A, Fassnacht M, Daffara F, Tauchmanova L, Conton PA, Rossetto R, Buci L, Sperone P, Grossrubatscher E, Reimondo G, Bollito E, Papotti M, Saeger W, Hahner S, Koschker AC, Arvat E, Ambrosi B, Loli P, Lombardi G, Mannelli M, Bruzzi P, Mantero F, Allolio B, Dogliotti L, Berruti A: Adjuvant mitotane treatment for adrenocortical carcinoma. N Engl J Med. 2007, 356: 2372-2380. 10.1056/NEJMoa063360.
European Public Assessment Report (EPAR) Signifor. 2012, EMEA/H/C/002052, London
Arnaldi G, Boscaro M: Pasireotide for the treatment of Cushing’s disease. Expert Opin Investig Drugs. 2010, 19: 889-898. 10.1517/13543784.2010.495943.
Colao A, Petersenn S, Newell-Price J, Findling JW, Gu F, Maldonado M, Schoenherr U, Mills D, Salgado LR, Biller BM: A 12-month phase 3 study of pasireotide in Cushing’s disease. N Engl J Med. 2012, 366: 914-924. 10.1056/NEJMoa1105743.
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO: Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005, 352: 987-996. 10.1056/NEJMoa043330.
Ortiz LD, Syro LV, Scheithauer BW, Rotondo F, Uribe H, Fadul CE, Horvath E, Kovacs K: Temozolomide in aggressive pituitary adenomas and carcinomas. Clinics (Sao Paulo). 2012, 67 (Suppl 1): 119-123. 10.6061/clinics/2012(Sup01)20.
Arenas C, Melian C, Perez-Alenza MD: Long-term survival of dogs with adrenal-dependent hyperadrenocorticism: a comparison between mitotane and twice daily trilostane treatment. J Vet Intern Med. 2014, 28 (2): 473-80. 10.1111/jvim.12303.
Stratakis CA: Cushing syndrome in pediatrics. Endocrinol Metab Clin North Am. 2012, 41: 793-803. 10.1016/j.ecl.2012.08.002.
Keil MF: Quality of life and other outcomes in children treated for Cushing syndrome. J Clin Endocrinol Metab. 2013, 98: 2667-2678. 10.1210/jc.2013-1123.
German Disease Management Guideline Therapy of Type 2 Diabetes - long recension. 1st edition, Version 2. 2013
German Disease Management Guideline Prophylaxis, Diagnosis and Treatment of Osteoporosis - long version. 2009
German Disease Management Guideline Depressive Disorder - long version. 1st edition, Version 3. 2012
German Disease Management Guideline Arterial hypertension. 2008
Acknowledgement
This systematic review was supported by intramural funding of the Federal Institute for Drugs and Medical Devices (BfArM). The support of Andreas Duda is gratefully acknowledged.
Disclaimer
The opinions mentioned throughout the following article are personal views of the authors and do not reflect an official position of the Federal Institute of Drugs and Medical Devices or an EMA-committee or working party, respectively.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors filed the manuscript, NE and VP performed a systematic search on clinical PK-parameter. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly credited. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Eckstein, N., Haas, B., Hass, M.D.S. et al. Systemic therapy of Cushing’s syndrome. Orphanet J Rare Dis 9, 122 (2014). https://doi.org/10.1186/s13023-014-0122-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-014-0122-8